
信息编译:抗内源性逆转录病毒的抗体促进肺癌免疫治疗
发表人:admin 发表时间:2023/7/18
原文
Antibodies against endogenous retroviruses promote lung cancer immunotherapy
Ng, K.W., Boumelha, J., Enfield, K.S.S. et al. Nature 616, 563–573 (2023).
https://doi.org/10.1038/s41586-023-05771-9
抗内源性逆转录病毒的抗体促进肺癌免疫治疗
摘要
B细胞经常在固体肿瘤的边缘被发现,作为异位淋巴器官的有组织的卵泡,称为三级淋巴结构(TLS)1,2。尽管TLS被发现与改善患者的生存率和对免疫检查点封锁(ICB)的反应有关,但这种关联的潜在机制仍然难以捉摸1,2。在这里,我们调查了TRACERx 421(通过治疗跟踪非小细胞肺癌进化)和其他肺癌队列患者的肺驻留B细胞反应,以及最近建立的肺腺癌免疫源性小鼠模型3。我们发现,人类和小鼠肺腺癌都会引起局部发芽中心反应和肿瘤结合抗体,并进一步确定内源性逆转录病毒(ERV)包膜糖蛋白为显性抗肿瘤抗体靶点。ERV靶向B细胞反应在人类和小鼠中都通过ICB以及小鼠模型中对KRAS(G12C)的靶向抑制而放大。ERV反应性抗体发挥抗肿瘤活性,延长了小鼠模型中的生存期,ERV表达预测了ICB在人类肺腺癌中的结果。最后,我们发现小鼠模型中有效的免疫治疗需要CXCL13依赖性的TLS形成。相反,治疗性CXCL13治疗增强抗肿瘤免疫力,并与ICB协同。我们的发现为TLS与免疫治疗反应的关联提供了可能的机制基础。
主要
尽管靶向疗法和免疫疗法取得了重大进展,但肺癌仍然是全球癌症相关死亡的主要原因。预测免疫检查点封锁(ICB)的反应仍然是一个挑战,尽管突变负担高,但70%的患者没有反应4。最近的研究已经确定,三级淋巴结构(TLS),肿瘤相邻基质中含有B细胞和T细胞的异位淋巴管器官,是几种癌症类型1,2的ICB反应的强预测因子,包括肺腺癌(LUAD)5,6,其中它们的存在和密度与更长的整体和无复发存活率独立相关1,2。然而,TLS、患者生存和免疫治疗反应之间关联的因果关系尚未确定1,2。
TLS包含类似于淋巴器官中发现的发芽中心(GC)的结构,其中B细胞在T滤泡辅助器(TFH)细胞的帮助下迭代突变其B细胞受体(BCR),这一过程增加了抗体反应的亲和力7。GC依赖于CXCL13-CXCR5趋化因子轴来组织B细胞卵泡,我们和其他人已将CXCL13确定为ICB反应的预测指标8,9,10。虽然TLS改善ICB反应的机制仍然不完全清楚,但对活性GC反应的要求意味着抗肿瘤抗体的贡献。抗肿瘤抗体经常在多种癌症类型中诱导,针对内部和肿瘤细胞表面抗原。这些肿瘤相关抗原(TAA)包括非突变分化抗原和共享肿瘤抗原,以及来自内源性逆转录病毒(ERV)的抗原11。虽然这种非突变抗原实际上是自身抗原,但它们在健康组织中的低表达和在癌症表观遗传景观中受限的上调分别导致癌症的不完全免疫耐受性和免疫原性12。癌症相关ERV抗原的免疫原性有助于发现这类TAA,以及三十年前小鼠癌细胞产生的传染性逆转录病毒13,14,15,但B细胞对这一类或其他TAA反应的后果或保护能力尚未完全描述。
在这里,我们评估了TLS、B细胞和抗肿瘤抗体对患者中治疗素和免疫疗法治疗的LUAD以及免疫疗法和靶向治疗的LUAD在新的小鼠模型3中的免疫保护的贡献,并发现肺驻留B细胞对ERV包膜糖蛋白反应的重要作用。
新的LUAD模型中的B细胞反应
为了研究B细胞和TLS在肿瘤进展和治疗反应中的作用,我们使用了一个新建立的LUAD模型,该模型基于KPAR细胞的移植和原位生长,该模型来自KrasLSL-G12D/+Trp53fl/fl(KP)背景3。免疫荧光染色显示B220+ B细胞聚集在KPAR肺肿瘤边缘周围,而CD3+ T细胞渗入肿瘤肿块(图1a)。在KPAR肿瘤附近发现了血管周成熟的TLS,T细胞和B细胞区域明显分离,后者包括基于Ki67染色的黑暗和光区,并表现出花生凝集素(PNA)阳性,符合活跃的GC反应(图1b,c和扩展数据图1a,b)。相比之下,带有常规非免疫性Trp53fl/flKrasLSL-G12D/+ KPB6肿瘤的肺3不含可识别的TLS(图1c和扩展数据图1a,b)。
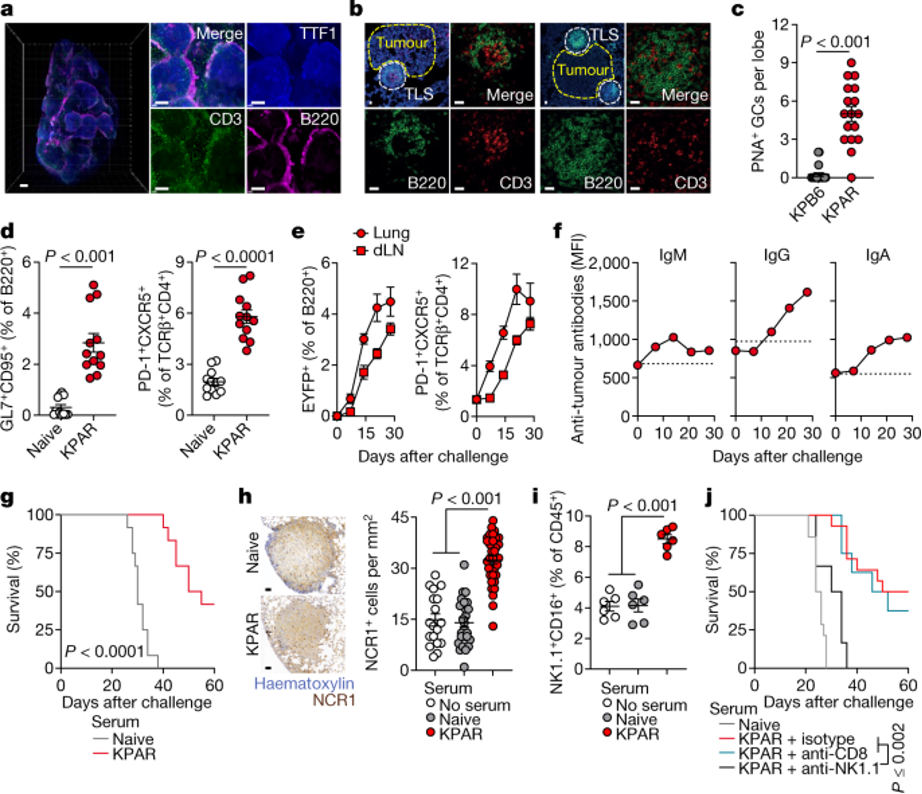 图1:小鼠LUAD中的B细胞反应。
图1:小鼠LUAD中的B细胞反应。
a,携带KPAR肿瘤的小鼠肺部B220(B细胞)、CD3(T细胞)和TTF1(肿瘤细胞)的免疫染色(鳞片棒,500微量)。五只小鼠的代表性图像。b、B220和CD3免疫荧光和DAPI染色在KPAR肿瘤肺部(鳞片条,20微分)。六只小鼠的代表性图像。c,通过组织化学对PPB6(n = 10)和KPAR(n = 4)含肿瘤肺叶的PNA+成熟TLS和GCs进行量化。d,B220+GL7+CD95+ GC B细胞和TCRβ+CD4+PD-1+CXCR5+ TFH细胞在天真和KPAR肿瘤的肺中的流动细胞进行定量(n = 12只小鼠,通过KPAR肺中的B220+EYFP+和TFH细胞的流式细胞仪进行时间过程定量,并从AicdaCreERT2Rosa26LSL-EYFP小鼠中排出淋巴结(dLNs)(n = 6小鼠从一个实验中每个时间点的小鼠)。f,KPAR血清的KPAR结合IgM、IgG和IgA的时间过程定量(n = 6)。虚线表示天真血清的平均染色强度。MFI,平均荧光强度。g,使用KPAR肿瘤携带或幼稚供体小鼠的集合血清治疗的KPAR受体小鼠的存活率(n =每组12小鼠,来自两个实验)。h,代表性图像(比例条,50 μm)和未经治疗或用天真或KPAR血清治疗的KPAR血清治疗的KPAR受体中肿瘤内NCR1+ NK细胞的量化(n =每组8只小鼠)。i,未经治疗或使用天真或KPAR血清治疗的KPAR接受者肺中NK1.1+CD16+ NK细胞的流式细胞测定定量(n =每组6只小鼠)。j,接受天真血清治疗的KPAR受体小鼠的存活率(n = 14)或使用KPAR血清和抗NK1.1(n = 6)、抗CD8(n = 8)或同型对照(n = 14)(来自两个实验)。c-f,h,i中的数据表示为均值± s.e.m。P值由c和d中的双面Mann-Whitney秩和测试(左)、d中的双面学生t测试(右)、带有Bonferroni校正的单向方差分析计算,用于h,i中的多重比较,以及g,j中的对数排名测试。
携带KPAR肿瘤的肺部流式细胞仪显示B220+GL7+CD95+ GC B细胞和TCRβ+CD4+PD-1+CXCR5+ TFH细胞明显升高,这与GC B细胞水平相关(图1d和扩展数据图1c)。相比之下,在带有KPB6肿瘤的肺部背景水平上发现了GC B和TFH细胞(扩展数据图1d)。这些数据表明,KPAR肿瘤,而不是KPB6肿瘤,刺激TLS形成和GC反应,正如在人类肺癌16,17中观察到的那样。
为了确认GC的形成,定义了成熟的TLS18,我们将KPAR细胞移植到AicdaCreERT2Rosa26LSL-EYFP(AID-EYFP)小鼠中,这些小鼠在AID酶表达后选择性地映射GC B细胞。Tamoxifen给药标记了75-85%的B220+GL7+CD95+ B细胞,经流式细胞仪评估(扩展数据图1e)。在KPAR挑战后的第7天,EYFP+细胞在B220+人群中可以检测到带有肿瘤的肺部和排出的淋巴结,并继续增加数量,直到终点,反映了TFH细胞动力学(图1e)。使用Ighg1CreRosa26LSL-五彩纸屑小鼠(扩展数据图1f)进一步确认了GC形成的动力学。
伴随着这些B细胞反应,来自KPAR挑战小鼠的端点血清,但不是天真或KPB6挑战小鼠,含有KPAR结合IgG和IgA抗体(扩展数据图2a,b)。KPAR结合的IgM抗体在KPAR挑战后的第14天达到峰值,此后下降,而类交换的IgG和IgA抗体与GC反应平行继续增加丰度(图1f)。
为了研究KPAR结合抗体的潜在抗肿瘤活性,我们将血清从KPAR挑战的捐赠者转移到KPAR挑战的二级接受者。与幼稚血清相比,KPAR血清的转移显著延长了接受者的生存期(图1g)。KPAR血清不会改变KPB6挑战接受者的存活率,KPB6血清不会影响KPAR挑战接受者的生存(扩展数据图2c,d)。
KPAR血清的抗肿瘤活性与肿瘤浸润自然杀伤者(NK)细胞数量的显著增加有关,这些细胞由NCR1表达进行组织学量化(图1h),以及表达CD16的NK细胞,即参与抗体依赖性细胞细胞毒性(ADCC)的Fc受体,由流式细胞仪量化(图1i)。支持NK细胞在介导KPAR血清抗肿瘤活性方面的作用,KPAR血清接受者NK细胞的枯竭消除了其保护作用(图1j)。相比之下,CD8+ T细胞的耗尽在这种环境中没有影响(图1j)。除了ADCC外,KPAR血清还引发了体外KPAR细胞的补体依赖性细胞毒性(CDC),这种毒性因血清热失活而减少(扩展数据图2e)。
这些结果表明,KPAR肿瘤,而不是KPB6肿瘤,诱导B细胞的招募和激活,并产生有效的抗肿瘤抗体。
抗肿瘤抗体靶向ERV
为了在KPAR模型中探究抗肿瘤抗体的特异性,我们首先考虑了非免疫性KPB6细胞不共享的假定细胞表面抗原。其中一类抗原是ERVs,包括内源性小鼠白血病病毒(MLV)包膜糖蛋白,其表达水平在KPAR中比在KPB6细胞中高得多3。我们发现,KPAR血清特异性染色小鼠癌细胞系,已知表达高水平的内源性MLV包膜糖蛋白15,但缺乏这种表达的小鼠肺癌细胞系或缺乏MLV包膜糖蛋白的人类肺癌细胞系(图2a和扩展数据图2f)。
 图2:小鼠LUAD中的抗ERV抗体。
图2:小鼠LUAD中的抗ERV抗体。
a,KPAR血清和83A25抗体与小鼠(B16、4T1、3LL、MC38、EL4、CTLL2)和人类(A549、HBEC)细胞系结合。该量表表示在朴素血清或同型对照组中的特定MFI增加。b,KPAR血清中M. dunni.KARV-和M. dunni结合IgM、IgG和IgA的量化(n =来自两个实验的6只小鼠)。虚线表示天真血清的MFI。c,来自天真或KPAR血清的KPAR结合IgG,被83A25或同型控制抗体阻断。五个独立复制的代表性直方图。d,接受83A25或同型对照或未经治疗野生型(WT)和Emv2−/−宿主治疗的KPAR肿瘤小鼠的生存(n =每组6只小鼠,一个实验)。e,KPAR和KPAR.eMLV−/−肿瘤小鼠的生存(n = 10小鼠,一个实验每组10小鼠)。f,GC B细胞、TFH细胞和KPAR结合IgG的量化在KPAR和KPAR.eMLV−/−肿瘤小鼠(n = 10小鼠每组)。g。g,KPAR治疗的KPAR治疗小鼠的生存率为抗PD-L1或异型对照(n =每组12小鼠,来自两个实验)。h,从KPAR肿瘤携带的GC B细胞和TFH细胞的肺中的GC B细胞的定量抗-PD-L1或同型对照治疗的小鼠 (n = 5 组5小鼠每组)。 i, KPAR-binding IgM, IgG和 IgGA来自治疗的小鼠血清治疗的小鼠的血清治疗抗-PD-PD-L1或异型控制 (n = 5小鼠每组)。 j, 接受者 KPAR挑战小鼠的生存治疗的KPAR挑战小鼠治疗抗PD-L1治疗 KPAR血清 (n = 20)或天真血清 (n = 18) (n = 18) (来自三个实验) k, BCR CDR3 clonotypes在抗PD-L1处理 KPAR肺中的频率 (n = 3,集合).l, J1KK和IgA同型结合 KPAR或M. dunni.KARV细胞. m,生存 KPAR肿瘤小鼠接受 J1KK IgA治疗的生存与 (n反NK1.1,或异型控制(n = 6)(来自一个实验)。b,f,h,i中的数据表示为均值± s.e.m。P值由b,f,h,i中的双面学生t测试和log排名测试ind,e,g,j,m计算。
与其他可移植小鼠细胞系15一样,KPAR细胞中内源性MLV包膜糖蛋白的表达升高可能是由于存在具有恢复传染性的MLV,该病毒来自复制缺陷的生态MLV(eMLV)原生病毒Emv2。事实上,我们通过通过Mus dunni细胞中的KPAR上清液分离出一种传染性MLV,我们称之为KPAR相关逆转录病毒(KARV),该上清剂与内源性MLV包膜特异性83A25抗体(扩展数据图2g)以及KPAR肿瘤小鼠的血清(图2b)产生强烈反应。
为了确定靶向KARV包膜糖蛋白的KPAR结合抗体的比例,我们用83A25预孵化了KPAR细胞,这导致内源性MLV包膜糖蛋白的内化19。这种处理废除了KPAR血清染色(图2c),将KARV确定为主要抗体靶点。
通过83A25的治疗,KPAR挑战的野生型小鼠的存活率显著延长,在缺乏Emv2的小鼠中,KPAR肿瘤生长延迟,这些小鼠对eMLV包膜糖蛋白20缺乏免疫耐受性(图2d)。此外,KPAR.eMLV-/-细胞中Cs9介导的Emb2衍生原病毒的缺失在皮下注射到野生型(但不是T和B细胞缺乏的接受者)后加速了肿瘤生长3。静脉注射后也获得了类似的结果,导致野生型受体的原位生长(图2e),同时由KPAR.eMLV-/-细胞引起的GC、TFH和抗肿瘤抗体反应显著减少(图2f)。因此,异常表达的ERV是自发产生的针对KPAR肿瘤的保护性抗肿瘤抗体的主要目标。
PD-L1封锁增强了反ERV响应
接下来,我们检查了GC反应和抗肿瘤抗体是否有助于该模型3中PD-1或PD-L1阻断的治疗效果。虽然遗传研究已经确定了PD-L1+ GC B细胞和PD-1+ TFH细胞在GC形成和功能中的相互作用的关键作用21,22,但阻断抗体对这些过程的影响尚未得到研究。我们首先通过给小鼠接种羊红细胞(SRBCs)来探索ICB在GC B细胞反应中的作用,这种作用与肿瘤生长的继发效应无关。与异型对照相比,使用抗PD-L1抗体治疗的小鼠在脾脏GC B细胞和TFH细胞以及增殖性暗区GC种群中有所增加(扩展数据图3a)。PD-L1封锁增加了单个GC的规模,但没有增加数量,这表明对先前存在响应的扩展而不是重新归纳的影响(扩展数据图3b)。PD-L1阻断比CTLA-4阻断更有效调节GC B细胞反应(扩展数据图3c),因此我们在随后的肿瘤实验中使用了抗PD-L1单药治疗。
PD-L1的阻断显著延长了KPAR挑战小鼠的生存期(图2g),类似于对其受体PD-1的阻断(参考3)。它还扩大了局部GC B细胞和TFH细胞反应(图2h),这些影响通过PD-1或CTLA-4阻断再现(扩展数据图4a)。PD-L1阻断显著增加了肿瘤结合IgG和IgA类交换抗体的滴度(图2i),这与PD-L1缺乏小鼠在模型抗原免疫后GC反应和抗体滴度的增加一致21。与免疫PD-L1缺乏小鼠21中产生的抗体亲和力降低相比,我们发现PD-L1阻断增加了而不是减少了抗体与KPAR细胞结合的总体贪婪(扩展数据图4b)。为了验证体内的抗体功能,我们测试了抗PD-L1治疗的捐赠者血清的治疗活性。我们首先确认这些血清不再含有抗PD-L1抗体(扩展数据图4c)。与异型治疗的KPAR捐赠者的血清接受者相比,捐赠小鼠的PD-L1封锁进一步延长了KPAR挑战的二级接受者的生存时间,而与来自天真捐赠者的血清接受者相比,后者又延长了生存时间(图2j),支持PD-L1封锁诱导的抗肿瘤抗体的功能。
来自抗PD-L1处理的KPAR挑战小鼠的血清显示IgG和IgA与KARV感染的M. dunni细胞的结合升高(扩展数据图4d),表明对这种ERV抗原的反应增强。为了直接询问特异性,我们从接受治疗的KPAR挑战小鼠的集合肺中分离的单个B细胞中测序了BCR。我们在这个池中发现了一个显性克隆,这里称为J1KK,由VH13-2段和IgA同种型编码,占所有Igh互补性确定区域3(CDR3)序列的20%(图2k)。重组J1KK单克隆抗体结合KPAR细胞的表面,以及KARV感染的M. dunni细胞的表面,指出KARV包膜糖蛋白为靶抗原(图2l)。J1KK结合的肽的质谱分析证实了它们的eMLV包膜起源(扩展数据图4e)。用J1KK和天真血清体外孵化KPAR细胞触发了CDC(扩展数据图4f),并以NK细胞依赖性的方式对KPAR挑战的小鼠进行IgA或IgG1版本的J1KK的体内治疗显著延长了生存期(图2m)。结合起来,这些数据确定了抗ERV抗体对未经治疗和ICB治疗的KPAR肿瘤反应的贡献。
靶向治疗中的B细胞反应
为了检查抗肿瘤B细胞反应是否有助于ICB以外的治疗效果,我们使用了靶向疗法,包括高度选择性的KRAS(G12C)抑制剂(G12Ci)23。我们首先将编码G12C替换的Kras突变引入KPAR细胞系(KPARG12C),并用于这些实验3。KPARG12C肿瘤的转录分析显示,在使用G12Ci MRTX-849治疗的肿瘤中,免疫球蛋白和GC B细胞相关基因转录有强烈的上调(图3a)。细胞反卷积表明G12Ci治疗的肿瘤中B细胞富集,经GC B细胞的流式细胞仪验证,并由TLS的组织学检测进一步支持(图3b-d)。
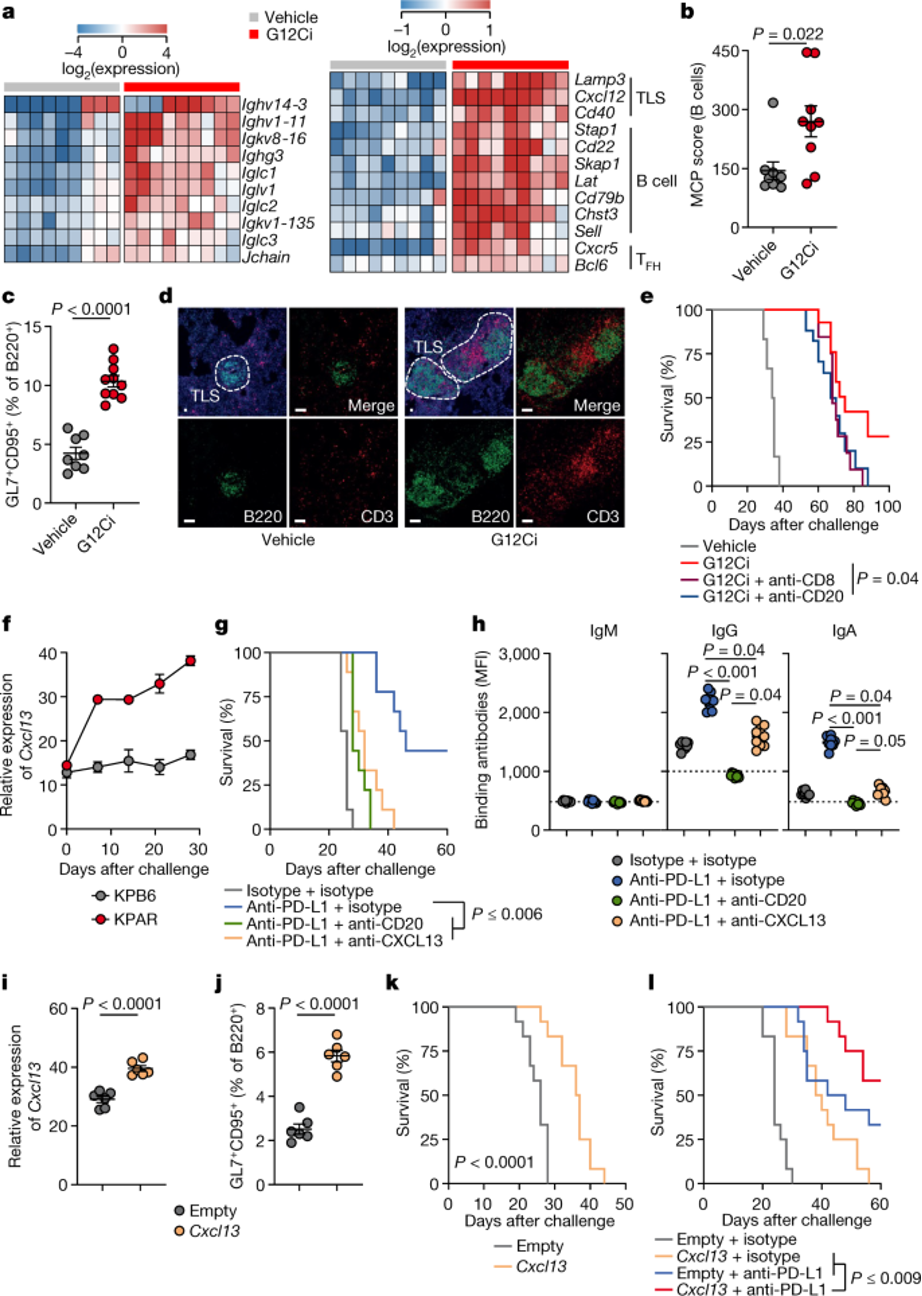 图3:LUAD疗法中的B细胞反应。
图3:LUAD疗法中的B细胞反应。
a,b,免疫球蛋白和TLS相关基因表达(a)和MCPCounter B细胞评分(b)在MRTX-849(G12Ci)或车辆对照治疗的KPAR肿瘤(n =每组9个小鼠,来自一个实验)。c,GC B细胞定量在G12Ci治疗(n = 10)或车辆处理(n = 8)的肺部来自KPAR挑战小鼠(来自一个实验)。d,B220(B细胞)和CD3(T细胞)免疫荧光和DAPI染色在G12Ci-和车辆控制治疗的肺部从KPAR挑战小鼠(量表条,20 μm)。四个个体小鼠的代表性图像. e, 生存 车辆控制处理 (n = 6)或 G12Ci-处理 (n = 16) KPAR-challenged小鼠和那些额外治疗的抗-CD20 (n = 17)或抗CD8 (n = 16)在 G12Ci治疗之前 (n = 16)之前 (来自 G12Ci治疗 (n = 16)。 f,时间过程量化 量化与逆转转录 (RT-qPCR) Cxcl13表达在 KPAR或 KPB6肺中的 Cxcl13表达 (n = 3每个时间点每个时间点从一个实验) g,h,h,生存 (g)和 KPAR-binding IgM, IgG和 IgG和 IgA水平在血清 (h)中治疗的 KPAR-PD-L1,抗-CD20和抗-CD20和抗 CXCL13或异型接受鼻内质粒编码Cxcl13或空载体控制治疗的KPAR挑战小鼠肺部的记录(n =两组6只小鼠)。j,接受鼻内质粒编码Cxcl13或空载体对照治疗的KPAR挑战小鼠肺部的GC B细胞量化(n =两组每组6只小鼠)。k,接受鼻内质粒编码Cxcl13或空载体控制治疗的KPAR挑战小鼠的生存(n =每组来自两个实验12只小鼠)。l,接受抗PD-PD-L1和Cxcl13或异型和空载体对照治疗的KPAR挑战小鼠的生存(n =每组12只小鼠在两次实验中)。b,c,f,h-j中的数据表示为均值± s.e.m。P值由b中的双面Mann-Whitney秩和测试、c、i、j中的双面学生t测试、带有Tukey校正的排名单向方差分析计算,用于h中三个治疗组之间的多重比较和e,例如k,l中的对数排名测试。
虽然KRAS(G12C)和有丝分裂原激活的蛋白激酶激酶(MEK)抑制剂通常被认为是同一治疗类别,但MEK在B细胞发育和激活中起着关键作用24。因此,MEK抑制剂(MEKi)曲美替尼减弱了GC和TFH对常规SRBC免疫的反应(扩展数据图5a)。相比之下,G12Ci没有影响GC或TFH对SRBC免疫的反应(扩展数据图5a),这表明其在KPARG12C挑战后的影响是肿瘤细胞的内在影响。在KPARG12C挑战的小鼠中,与MEKi或车辆控制相比,G12Ci治疗增强了GC和TFH反应,以及抗肿瘤IgG和IgA抗体水平(扩展数据图5b,c)。此外,使用MEKi(而不是G12Ci)治疗对抗肿瘤抗体的贪婪产生了不利影响(扩展数据图5d)。这些数据表明,KRAS(G12C)的肿瘤细胞特异性抑制促进了KPAR模型中的抗肿瘤B细胞反应,但无处不在的MEK抑制阻碍了抗肿瘤B细胞反应。为了探索B细胞是否积极促进对G12Ci的持久反应,我们在G12Ci之前用CD20耗竭抗体治疗了小鼠。B细胞耗尽增加了G12Ci治疗的KPARG12C挑战小鼠的复发率,随后降低了生存率,类似于CD8+ T细胞耗尽;然而,这种影响没有达到统计学意义(图3e),表明G12Ci可能有助于免疫记忆防止肿瘤复发。
CXCL13疗法与ICB协同
为了量化TLS和抗肿瘤B细胞反应对KPAR肿瘤的抗性的贡献和要求,我们抑制了淋巴结构组织趋化因子CXCL13。KPAR后,Cxcl13在小鼠肺部的表达增加,但没有增加KPB6,挑战(图3f),这意味着CXCL13在随后的局部GC反应中的作用。为了测试这一点,我们使用了CXCL13阻断方案,该方案之前发现可以消除肺部的GC反应,而不是甲型流感病毒(IAV)感染期间排出的淋巴结25。因此,CXCL13阻断减少了抗PD-L1治疗的KPAR挑战小鼠肺部的GC B细胞反应,但没有减少流肺结中的淋巴结(扩展数据图5e),并否定了ICB的治疗效果(图3g)。这些影响伴随着抗肿瘤IgG和IgA抗体滴度的降低(图3h)。作为对照,使用CD20耗竭抗体治疗的抗PD-L1治疗的KPAR挑战小鼠在系统性地失去了GC B细胞反应(扩展数据图5e)和完全失去抗肿瘤抗体(图3h),但对ICB不敏感,类似于使用CXCL13阻断抗体(图3g)治疗的小鼠。相比之下,仅抗CD20或抗CXCL13抗体对没有额外接受ICB的KPAR挑战小鼠的生存影响微乎其微(扩展数据图5f)。这些发现支持了对CXCL13编排的肺GC B细胞和抗肿瘤抗体反应的直接要求,这些反应支持了有利的ICB结果。他们还建议,CXCL13治疗可以进一步改善KPAR模型中ICB的抗肿瘤作用,正如结直肠癌和卵巢鼠癌模型的实验所表明的那样26,27。为了检查CXCL13的治疗作用,我们通过鼻内给药编码Cxcl13的哺乳动物表达载体与阳离子脂质GL67复合来治疗KPAR挑战的小鼠。与空载体相比,这种治疗增加了KPAR肿瘤肺部的Cxcl13表达(图3i)。它还增加了GC B细胞对KPAR挑战的反应,并显著延长了接受者的存活时间(图3j,k)。此外,与任何一种单一疗法相比,CXCL13和抗PD-L1的结合进一步延长了生存期(图3l),突出了基于吸入的免疫调节与ICB协同的潜力。
LUAD患者的B细胞反应
为了调查小鼠模型所建议的体液免疫在确定人类肺癌亚型结果中的作用,我们比较了TRACERx 421队列中LUAD和肺鳞状细胞癌(LUSC)治疗素患者中的转录组B细胞和TLS特征。与来自邻近组织的正常肺样本相比,LUAD和LUSC肿瘤区域的TLS转录特征都减少了,当比较配对样本时,LUSC的这种减少更强(扩展数据图6a,b)。相比之下,与最近的一份报告6一致,两种亚型的B细胞特征都显著升高,但LUAD的LUAD程度高于LUSC(扩展数据图6a,b)。TLS和B细胞特征与肿瘤纯度成反比(扩展数据图6c),这意味着肿瘤组织稀释了正常肺部中存在的特征。事实上,其他指标,包括BCR剧目多样性、IgG频率和CXCL13表达以及组织学TLS检测,表明LUAD和LUSC中B细胞反应的诱导(扩展数据图6a,d)。
B细胞标记CD79A、CD19和MS4A1(编码CD20)的较高表达与LUAD的TRACCERx患者的预后更好,而不是LUSC,在TCGA(癌症基因组图谱)中独立,LUAD患者的预后更好,而不是LUSC(扩展数据图7a,b)。此外,高CXCL13表达与患有LUAD的TRACERx患者的无病生存率提高有关,但不是LUSC(扩展数据图7a),与TCGA患者LUAD患者的总体生存率提高有关,但不是LUSC(扩展数据图8a)。在TCGA队列中,高CXCL13表达是肿瘤类型的预后,其中报告了TLS密度和对ICB反应之间的关联1,2,其预后值与整体表达水平无关(扩展数据图8a,b)。
LUAD患者的ERV反应抗体
我们的结果表明,TLS和B细胞反应可能具有保护作用,特别是在LUAD中。然而,B细胞和TLS特征以及CXCL13表达,正如预期的那样,它们彼此密切相关,也与细胞毒性CD8+ T细胞和NK细胞特征显著相关(扩展数据图8c),与其他癌症类型1,2的发现一致。为了探索抗肿瘤B细胞反应对TLS和B细胞特征与LUAD患者生存的观察关系的可能直接贡献,而不是这是CD8+ T细胞反应的反映,我们调查了B细胞对TAA的反应性。根据之前的报告6,总肿瘤突变负担(TMB)与LUAD患者单个肿瘤区域的BCR剧目多样性和IgG频率显著相关,但与TLS或B细胞特征无关(扩展数据图9a)。同样,没有观察到吸烟状态或TP53、EGFR或KRAS突变的显著影响,但该队列中具有亚克隆TP53突变的肿瘤区域的TLS和B细胞特征减少除外(扩展数据图9b),尽管最近报告了有吸烟史的LUAD患者血浆细胞明显升高6。
接下来,我们检查了非突变TAA,重点是ERV包膜糖蛋白。我们首先检查了已知的人类ERV(HERV)位点可能编码包膜糖蛋白的转录。在37个这样的HERV位点(补充表1)中,34个在TCGA和TRACERx LUAD和LUSC中表现出可检测的表达(扩展数据图10a)。其中,1q22号染色体上的HERV-K(HML-2)亲病毒,这里称为ERVK-7(也称为HERV-K102),以及7q11.21染色体上的HERV-R亲病毒,这里称为ERV3-1,是两个LUAD队列中表达最多的位点(扩展数据图10a)。这两个位点也在LUSC中表达,LUSC在4q12号染色体上还表达了高水平的MER34原病毒,这里称为ERVMER34-1(编码内源性逆转录病毒包膜糖蛋白HEMO28)(扩展数据图10a)。
为了评估这些HERV在肿瘤类型中的表达,我们比较了泛组织TCGA和基因型组织表达(GTEx)数据集(31种癌症和33种健康组织类型)。ERV3-1和ERVMER34-1在几个健康组织中以高水平表达,包括在造血室和肾脏中(扩展数据图10b),如最近描述28。虽然ERVK-7在非恶性肺中表达,但在TCGA和TRACERx队列中,LUAD患者的表达明显上调,但在LUSC患者中没有上调(图4a和扩展数据图10b)。此外,对TRACERx患者的多区域肿瘤样本和配对正常组织的比较显示,ERVK-7表达的异质性相当大,但患者内部有限(图4b)。
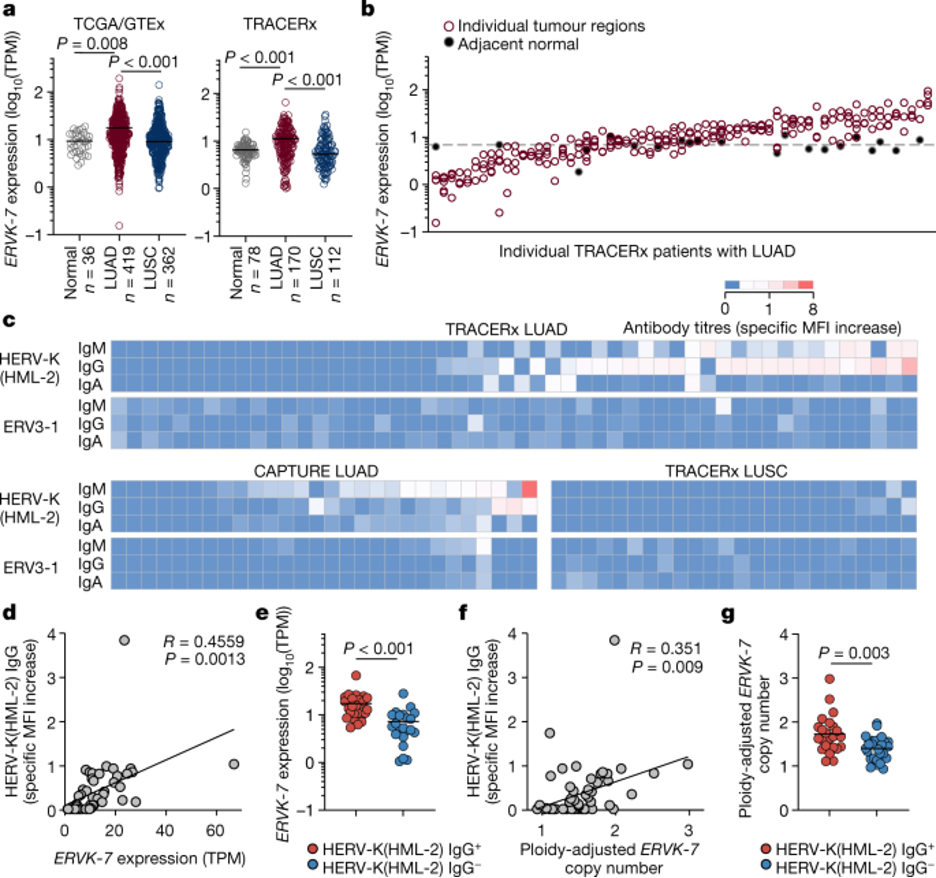 图4:LUAD和LUSC患者的抗HERV抗体。
图4:LUAD和LUSC患者的抗HERV抗体。
a,在TCGA LUAD(n = 419)和LUSC(n = 362)样本和GTEx健康肺样本(n = 36)(左)和TRACERx LUAD(n = 170)、LUSC(n = 112)和相邻正常组织(n = 78)样本(右)中每百万转录物中ERVK-7的表达。对于TRACERx患者,肿瘤值代表所有单个肿瘤区域的平均表达。b,ERVK-7在患有LUAD的TRACERx患者的多区域样本中的表达(n = 63名患者,至少有三个区域的数据)。填充符号和虚线分别代表单个配对的正常肺组织样本和所有正常肺组织样本的平均表达。c,通过流式细胞仪对患有LUAD(n = 52)和LUSC(n = 24)的TRACERx患者以及LUAD(n = 28)的TRACERx血浆或血清中的HERV-K(HML-2)和ERV3-1包膜结合抗体进行定量。对照细胞的特异性MFI增加值用量表表示。d,e,HERV-K(HML-2)包膜反应性IgG滴度和ERVK-7 mRNA表达(n = 47)(d)和ERVK-7 mRNA表达的相关性,在具有(HERV-K(HML-2)IgG+,n = 25)和没有(HERV-K(HML-2)IgG-2)IgG−,n = 22)HERV-反应抗体(e)的TRACERx患者中,HERV-K(HML-2)包膜反应抗体(h-K(HML-2)包膜反应抗体(n = 53)(f)和倍体调整的ERV-2)的ERV-K-7复制数的相关性(HML-2)与(HML-gG+,n = 23)的TRACERy轴代表每个患者在单个肿瘤区域的最大拷贝数。a和b中的符号分别代表单个患者和单个区域,P值通过单向方差分析计算,邓恩校正在a和双面曼-惠特尼秩和测试中进行多次比较,例如;R和P值使用d,f中的线性回归计算。
总体ERVK-7表达与细胞毒性CD8+ T细胞和NK细胞的转录特征以及IgG频率最密切相关,但与TLS或B细胞特征没有关系(扩展数据图11a)。这是可以预期的,因为ERVK-7位点的重叠转录物只有一小部分对应于包膜糖蛋白mRNA,其余对应于其他病毒蛋白的基因组RNA或mRNA。此外,ERVK-7是几个可检测表达的HERV-K(HML-2)位点之一,可能编码高度相似的包膜糖蛋白(95-98%的氨基酸特性)。LUAD组织微阵列中HERV-K(HML-2)包膜糖蛋白的染色表明,该蛋白质在患者中确实以可变水平表达,在肿瘤中表达水平高于相邻的正常细胞(扩展数据图11b),增加了它刺激B细胞反应的可能性。
接下来,我们使用之前描述的流式细胞仪检测29,对手术前TRACERx患者血浆样本进行了ERV包络糖蛋白反应抗体的筛查。尽管两种组织学亚型都存在转录物表达,但在45%的LUAD患者中检测到抗体,主要是IgG和IgM,与祖先HERV-K(HML-2)包膜蛋白反应,没有检测到任何LUSC患者。在LUAD30患者的验证队列中也检测到抗HERV-K(HML-2)抗体,频率为28%(图4c)。相比之下,除了一名LUAD患者外,所有针对ERV3-1包膜蛋白的抗体都无法检测到。这表明HERV-K(HML-2)包膜糖蛋白可以刺激体液反应,优先在LUAD中。
在TRACERx LUAD队列中,ERVK-7转录水平与HERV-K(HML-2)包膜反应性IgG抗体(图4d,e)的滴度显著相关,支持了一个模型,其中ERVK-7的转录激活打破了对HERV-K(HML-2)包膜糖蛋白的免疫耐受性。因此,我们调查了升高的ERVK-7转录的潜在机制。这种原病毒最近已被证明在其他情况下对表观遗传变化和转录因子SOX2有反应31。然而,在TCGA LUAD样本中,ERVK-7转录与全局甲基化或SOX2表达之间没有发现相关性(扩展数据图11c),尽管这种分析不排除局部表观遗传变化的影响。作为替代方案,我们考虑了在LUAD进化32期间经常发生的1q22号染色体的扩增可能通过创建额外的ERVK-7基因组副本导致ERVK-7表达升高。根据这一假设,我们发现ERVK-7表达与TRACERx LUAD队列中的倍体调整ERVK-7拷贝数以及TCGA LUAD队列中ERVK-7基因组位点的平均拷贝数相关(扩展数据图11d)。此外,患有LUAD的TRACERx患者的抗HERV-K(HML-2)包膜抗体的滴度与倍体调整的ERVK-7拷贝数显著相关(图4f,g)。总的来说,这些数据表明,在相当一部分LUAD患者中存在HERV-K(HML-2)包膜反应抗体,这可能是由ERVK-7转录增加引起的,而ERVK-7转录又得到了1q22染色体扩增的帮助。
ICB增强人类抗ERV抗体
为了评估区域淋巴结对TLS BCR剧目中的相对贡献,我们寻找特异于患有LUAD的TRACERx患者肿瘤区域的B细胞克隆扩张。在患有LUAD的TRACERx患者CROK0035中,一个IgG1类交换重链和一个轻链(此处称为103-K7)分别占肿瘤区域1中所有生产性BCR的32.4%和25.3%,而来自配对正常肺组织的BCR缺乏显性克隆(图5a),表明肿瘤特异性克隆扩张。与生殖系基因段相比,103-K7重链和轻链重排分别具有七种氨基酸和一种氨基酸替换,在另外两名患者中也发现了这种组合,频率要低得多。这些也在患者CROK0035的肿瘤区域2中以较低的频率发现,但在第三个肿瘤区域、淋巴结转移或配对的正常肺组织中没有发现(图5b)。相反,在淋巴结转移和所有三个肿瘤区域中以高频发现非突变的103-K7前体,但在配对的正常肺组织中没有发现(图5b)。虽然这种抗体克隆的确切特异性仍有待确定,但这些结果表明,103-K7前体起源于淋巴结,并播种了所有采样的肿瘤区域,但随后在肿瘤区域1中进一步进行类切换、超突变和克隆扩张。
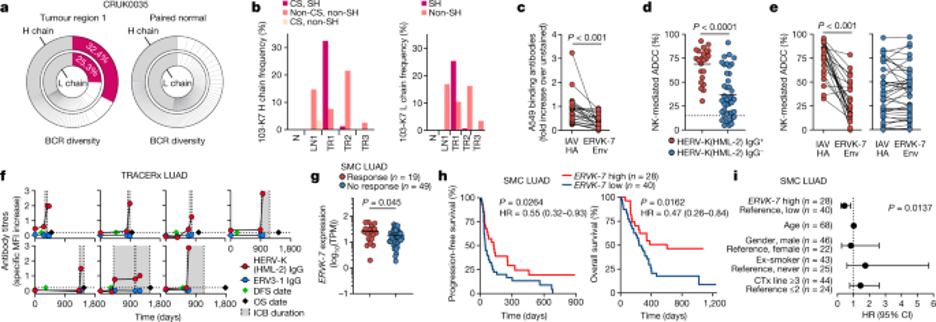 图5:LUAD患者的HERV-K(HML-2)反应抗体。
图5:LUAD患者的HERV-K(HML-2)反应抗体。
a,频率所有重 (H)和轻 (L)链 BCR CDR3重排列在肿瘤区域 1和配对正常肺组织从 TRACERx患者 CRUK0035与LUAD. b,重链和轻链频率 103-K7 clonotype,非类切换 (非CS)和非躯体超突变(非SH)前体,以及类切换和非躯体超突变前体,在三个独立的肿瘤区域 (TR1-TR3),淋巴结转移 (LN1)和配对正常肺组织 (N)从患者 CRUK0035. c–e, A549 binding (c)和 A549 ADCC (d,e)从 TRACERx患者与 LUAD with (IgG+, n = 23)或无 (IgG−, n = 41) HERV-K-K(HML-2) 包络-2) 包络-抗体无 (或添加(c,e)重组ERVK-7包膜蛋白或IAV血凝素(IAV HA)。 f,HERV-K(HML-2)和ERV3-1包络反应IgG滴度在LUAD患者在 ICB之前和期间(灰色),根据手术后时间(第0天)(虚线,检测极限;DFS,无病生存;OS,总生存率)。g,根据 ICB治疗反应, LUAD的SMC患者的ERVK-7 mRNA水平。 h,根据治疗前ERVK-7表达水平,在 LUAD患者中,根据治疗前ERVK-7表达水平,总体生存风险比(HRs)在ICB治疗后(CTx,化疗)。i中的误差条表示95%的置信区间(CI)。c-g中的符号代表单个患者,c和e中的线条连接同一患者的值。P值由c中的Wilcoxon有符号排名测试、d中的双面学生t测试、e中的双面配对学生t测试、g中的双面Mann-Whitney秩和测试、h中的对数排名测试和i中的Cox比例危害回归计算。
为了探究HERV-K(HML-2)包膜反应抗体在LUAD中的功能相关性,我们首先估计了它们所构成的整体抗肿瘤反应的比例。HERV-K(HML-2)包膜反应抗体的患者血浆也染色了A549细胞,与对照IAV血凝素(图5c)相比,通过添加可溶性重组ERVK-7包膜糖蛋白,这种染色平均减少了50%(-30%至97%)。HERV-K(HML-2)包膜反应抗体的LUAD患者的血浆介导的ADCC对A549目标的效率明显高于没有HERV-K(HML-2)包膜反应抗体(图5d)。此外,添加可溶性重组ERVK-7包膜糖蛋白平均抑制55%(-15%至100%)由血浆介导的ADCC与HERV-K(HML-2)包膜反应抗体,而没有HERV-K(HML-2)包膜反应抗体的血浆活性(可能针对替代共享肿瘤抗原)没有受到影响(图5e)。这些结果表明,HERV-K(HML-2)包膜靶向抗体占抗肿瘤体液反应的很大一部分,在罕见的情况下,其整体。此外,HERV-K(HML-2)包膜靶向抗体可以介导有效的抗肿瘤作用,这与其他系统中的发现一致33。
为了探索HERV-K(HML-2)包膜反应抗体在免疫治疗期间是否有助于抗肿瘤免疫,我们监测了七名接受ICB的TRACERx LUAD患者的滴度。启动ICB治疗后,所有七名患者的HERV-K(HML-2)包膜反应抗体滴度迅速上升,与之前的滴度或之前的非ICB治疗无关(图5f)。相比之下,ERV3-1反应性抗体的滴度仍然无法检测到(图5f),这表明ICB在促进HERV-K(HML-2)包膜糖蛋白的抗体反应方面具有特殊作用。虽然ICB停止后的存活率与HERV-K(HML-2)包膜反应抗体滴度的上升(R = 0.770,P = 0.042)呈正相关,但该ICB治疗队列的小型无法根据结果对抗体水平进行全面比较。因此,在之前描述的来自三星医疗中心(SMC)的更大群LUAD34患者中,我们检查了ERVK-7可能参与ICB治疗结果,其中提供了RNA测序(RNA-seq)数据。该队列中编码逆转录病毒包膜糖蛋白的HERV位点的表达与TCGA和TRACERx队列中的表达相似,ERVK-7是表达度最高的亲病毒(扩展数据图11e)。与ICB未经治疗的LUAD TRACERx患者类似,LUAD的SMC患者的ERVK-7表达与CD8+ T细胞特征显著相关(扩展数据图11f)。值得注意的是,在对ICB治疗有反应的LUAD的SMC患者中,治疗前ERVK-7表达水平高于没有反应的患者(图5g)。此外,虽然在未接受ICB治疗的患者中没有预后,但较高的治疗前ERVK-7表达与ICB治疗后更好的无进展和总体生存率显著相关,因此可以预测结果,与年龄、性别、吸烟状态和之前的非ICB治疗无关(图5h,i)。这些结果支持了ERVK-7表达的可能参与,以及随之而来的HERV-K(HML-2)包膜靶向抗体反应在抗肿瘤免疫中的参与,这是成功ICB治疗的基础。
讨论
总的来说,我们的发现表明,局部和系统性抗肿瘤B细胞反应可能会在小鼠和人类LUAD中发展,并通过产生肿瘤结合抗体来促进抗肿瘤免疫。这些B细胞和抗体反应可以靶向ERV包膜糖蛋白,并通过免疫疗法得到促进,为在人类中观察到的TLS和ICB反应之间的联系提供了一个潜在的机制。这些发现与诱变免疫原性乳腺癌模型中的类似发现一致,其中B细胞和TFH反应在ICB35后得到增强,并为TLS和肺癌免疫治疗反应之间新出现的联系提供了进一步支持1,2,18,35。ICB促进抗肿瘤抗体反应也表明PD-1/PD-L1定向免疫疗法对自身和外来抗原的体液反应有更广泛的影响,如使用模型抗原和人类中所示,据报道,ICB提高了循环CXCL13水平和季节性流感疫苗接种的抗体反应36。除了ICB,TLS的形成与新辅助化疗和靶向HER2治疗的反应相关37,38,反映了我们的G12Ci数据,并表明TLS在肿瘤细胞靶向治疗中可能具有意想不到的作用。与此形成鲜明对比的是,针对肿瘤和正常细胞的疗法,如MEK抑制,可能会对诱导肿瘤的适应性免疫反应产生不利影响。这些发现表明,将MEK抑制剂与肺癌中的KRAS(G12C)抑制剂相结合,或黑色素瘤中的BRAFV600E抑制剂,可能会损害抗肿瘤免疫反应,从而限制ICB组合的治疗效果和可能益处。
B细胞的一个关键功能是产生抗体。抗肿瘤B细胞和抗体反应通常针对非突变、过度表达的自身抗原,并受到一定程度的免疫耐受性11,39。长期以来,ERVs作为肿瘤抗原的作用在小鼠模型中被描述,首先是与起源于C57BL/6小鼠的黑色素瘤反应的单克隆抗体,该抗体被发现是这些黑色素瘤共享的eMLV的包膜糖蛋白特异性的40。具有恢复传染性的MLV经常在小鼠癌症模型中出现,通常是通过有缺陷的eMLV前体之间的重组,并导致小鼠肿瘤细胞中MLV抗原的表达增加和免疫原性增加15,20。虽然人类尚未恢复内源性逆转录病毒感染性,但HERV表达的转录上调仍可能允许在癌症患者中诱导HERV特异性抗体,主要是针对最近内原化HERV-K(HML-2)组的成员33,41,42。尽管最近在SOX2表达细胞31中提出了HERV-K(HML-2)亲病毒的动员,包括ERVK-7,但在这里,我们提供了证据,证明ERVK-7副本可以放大的新机制,即其染色体位点的扩增。HERV-K(HML-2)包膜糖蛋白的表达主要由ERVK-7在LUAD中表达是基于转录证据的研究。然而,非常相似,因此可能是抗体交叉反应的HERV-K(HML-2)包膜糖蛋白由几种原病毒编码,其中一些在人类中是插入多态的。因此,确定每个原病毒对健康和转化细胞中整体HERV-K(HML-2)包膜糖蛋白抗原池的贡献可能很重要。
HERV-K(HML-2)包膜糖蛋白的抗体在人类乳腺癌异种移植模型中表现出抗肿瘤活性,独立于适应性免疫细胞33。此外,据报道,治疗前HERV-K的表达可以预测胰腺癌和结直肠癌患者对联合免疫治疗和放疗的反应,并在治疗后患者中进一步上调,尽管没有评估HERV-K在肿瘤细胞上的蛋白质表达,也没有评估特异性抗体43。在SARS-CoV-2感染29后,以及在一定比例的健康个体和系统性红斑狼疮(SLE)患者中,也检测到HERV-K(HML-2)包膜反应抗体44。虽然健康捐赠者和SLE患者之间的滴度相似,但它们仅与后者44的干扰素活性相关,这表明HERV-K(HML-2)包膜反应抗体可能具有需要进一步研究的功能活性。
总体而言,我们的数据支持以下概念,即局部和全身B细胞反应通过产生保护性抗体来促进治疗反应,并将ERV包膜糖蛋白确定为相关的肿瘤抗原。了解B细胞的肿瘤和亚型特异性作用对于告知使用靶向B细胞扩张作为预测肿瘤对免疫治疗的反应甚至致敏的机制至关重要。
方法
小鼠菌株
C57BL/6J野生型小鼠,Aicdatm1.1(cre/ERT2)Crey(AicdaCreERT2)小鼠45,Ighg1tm1(cre)Cgn(Ighg1Cre)小鼠46,Gt(ROSA)26Sor tm1(EYFP)Cos(Rosa26LSL-EYFP)小鼠47,Gt(ROSA)26Sor tm1(CAG-Brainbow2.1)Cle(Rosa26LSL-Confetti)小鼠48和Emv2缺陷小鼠20之前已被描述过,并保存在弗朗西斯·克里克研究所生物研究设施,其遗传背景为C57BL/6J。小鼠被安置在恒温(21-25 °C)和湿度(50-60%)的通风笼子里,在标准的12小时光/12小时暗循环和特定无病原体条件下。所有实验都使用8至12周大的雄性或雌性小鼠,随机分配给年龄和性别匹配的治疗组,生存分析被盲目化。动物数量是根据我们实验室肿瘤生长的试点研究估算的。所有实验都得到了弗朗西斯·克里克研究所伦理委员会的批准,并根据当地指导方针和英国内政部根据1986年《动物科学程序法》(ASPA)的规定进行。
细胞系
KPAR细胞是来自Trp53fl/flKrasLSL-G12D/+背景的KPAR1.3系,正如最近描述的3。KPARG12C细胞是KPAR1.3线3的KRAS(G12C)表达衍生物。
HEK293T.ERV3-1env和HEK293T.HERV-K(HML-2)env细胞如前所述29。简而言之,HEK293T.HERV-K(HML-2)env细胞是通过HEK293T细胞的逆转录病毒转导产生的,其载体编码HERV-K113包膜糖蛋白49的假定祖先蛋白序列的密码子优化版本,由N.Bannert和GFP被内部核糖体进入点(IRES)隔开。HEK293T.ERV3-1env细胞同样由逆转录病毒转导产生,载体编码ERV3-1包膜糖蛋白(NCBI参考序列:NM_001007253.4)和GFP由IRES分离。KPAR、KPARG12C、KPB6、M. dunni、HEK293T、HEK293T.ERV3-1env、HEK293T.HERV-K(HML-2)env、EL4、CTLL2、B16、4T1、3LL、MC38、A549、NK92和HBEC细胞从中获取并验证为无支原体,并通过弗朗西斯·克里克研究所细胞服务设施通过DNA指纹对人类细胞系进行了额外验证。细胞在DMEM(Thermo Fisher)、RPMI(Thermo Fisher)或IMDM(Sigma-Aldrich)中培养,并辅以FBS(10%;Thermo Fisher)、L-谷氨酰胺(2 mM;Thermo Fisher)、青霉素(100 U ml-1;Thermo Fisher)和链霉素(100 μg ml-1;Thermo Fisher)。M. dunni.KARV细胞是通过培养M. dunni细胞产生的,这些细胞允许所有描述的内源性eMLV,在KPAR细胞的条件培养基中产生,并通过染色83A25单克隆抗体进行验证。
肿瘤模型和免疫接种
对于原位肺肿瘤模型,将1.5 × 105 KPAR、1.5 × 105 KPARG12C或1 × 105 KPB6细胞静脉注射到尾静脉。小鼠每周称三次体重,并在达到15%的减肥人道终点时被杀死。在免疫实验中,小鼠用2×108个SRBC(Fitzgerald Industries)进行腹腔内免疫。
对于抗体治疗,200微克抗PD-L1(10F.9G2,BioXCell)、抗PD-1(RMP1-14,BioXCell)、抗CTLA-4(9H10,BioXCell)、抗CXCL13(143614,研发系统)、抗NK1.1(PK136,BioXCell)、抗CD8(53-6.7,BioXCell)、抗eMLV Env(83A25,内部)、抗KARV Env(J1KK,内部)或其各自的同型对照组每周在腹腔内注射两次。对于B细胞耗竭实验,小鼠接受单次静脉注射250微克抗CD20(SA271G2,BioLegend)的治疗。对于血清转移实验,通过末端出血从KPAR肿瘤小鼠中收集血清,在56°C下热灭活10分钟,并在-20°C下储存。从第7天开始,每周两次从10只小鼠中注射接受肿瘤的100微升血清。从第7天开始,每周两次用抗NK1.1、抗CD8或同型对照抗体治疗图1j和2m中的小鼠。
对于KRAS或MEK通路抑制剂实验,一旦通过微计算断层扫描(CT)检测到肿瘤,就会开始治疗。通过吸入异氟醚对小鼠进行麻醉,并使用量子GX2微CT成像系统(PerkinElmer)扫描,各向同性像素大小为50微米。然后,通过口服给药给药50毫克kg-1 MRTX-849(MedChem Express),3 mg kg-1 trametinib(LC实验室)或车辆。在图例中所示的持续时间内,小鼠每天接受抑制剂。图3a-d中的小鼠在检测到肿瘤后6天内每天接受抑制剂或车辆对照治疗。图3e中出现KPAR肺肿瘤的小鼠在2周每日G12Ci治疗开始前1天使用抗CD20、抗CD8或同型对照抗体进行治疗,并监测其存活率,直到终点。对于接受抗CD8治疗的小鼠,在G12Ci终止后,每周注射两次,治疗继续进行。
肺基因转移
小鼠Cxcl13 cDNA ORF(NM_018866.2)被合成并克隆到pcDNA3.1哺乳动物表达载体(Genscript)中。为了制备GL67脂质粒,用1.21 mM GL67脂质体(Genzyme)培养1.6 mg ml–1 pcDNA3.1-Cxcl13或pcDNA3.1作为空载体控制,以给出最终的1:4摩尔比。通过吸入异氟醚对小鼠进行麻醉,并每周两次在鼻内注射20微升的GL67-质粒复合物。
流式细胞仪
肺部用20毫升的冷PBS灌注,切成小块,在37°C的PBS中用1毫克毫升-1胶原酶(Thermo Fisher)和50U毫升-1 DNase I(生命技术)孵化30分钟。样品通过70微米的尼龙过滤器过滤,在FACS缓冲液中重新悬浮(PBS中2%的FCS和0.05%叠氮化钠)之前,使用0.83%的氯化铵对红细胞进行解。样品在室温下用荧光标记抗体染色30分钟,抗体对CD45(BioLegend,30-F11)、B220(BioLegend,RA3-6B2)、GL7(BioLegend,GL7)、CD95(BioLegend,SA362F7)、CXCR4(BioLegend,L276F12)、CD86(BioLegend,GL-1)、TCRβ(BioLegend,H57-597)、CD4(BioLegend,GK1.5)、PD-1(BioLegend,29F.1A12)或CXCR5(BioLegend,L138D7)或无标签的抗eMLV Env(83A25,内部)、抗老鼠IgG(BioLegend,Poly4060)、抗老鼠IgA(Southern Biotech,11-44-2)、抗老鼠IgM样本在运行BD FACSDiva v.8.0的LSR Fortessa或运行Bio-Rad Everest v.2.4的Ze5分析仪上运行,并使用FlowJo v.10进行分析。用于识别不同细胞类型的门控策略如扩展数据图12a所示。
组织学和二维免疫荧光
含肿瘤的肺在10%的中性缓冲福尔马林(Sigma-Aldrich)中固定24小时,并转移到70%的乙醇或在OCT中冷冻。TRACERx卡扣冷冻的区域样本在首次服用足够的材料进行DNA和RNA测序后,被加工成固定的福尔马林、石蜡嵌入(FFPE)块。然后,通过从区域FFPE块中获取1.5毫米的核心来创建组织微阵列。固定组织嵌入石蜡中,4微米的部分安装在幻灯片上。使用自动组织-Tek Prisma幻灯片染色器进行血红素和伊红染色。对于免疫组织化学染色,石蜡嵌入部分在柠檬酸钠缓冲液(pH 6.0)中煮沸15分钟,然后用抗B220(1:250;RA3-6B2,BD Biosciences)、抗CD8(1:250;4SM15,Thermo Fisher)、抗Ki67(1:250;MIB-1,Agilent)、抗NCR1(1:250;ab233558,Abcam)、PNA(1:250;B1075,Vector Laboratories)或抗ERVK-7(1:250;PA5-49515,Thermo Fisher)孵化1小时。使用辣根过氧化物酶(HRP)共轭抗大鼠IgG(1:1,000;多克隆;Thermo Fisher,31470)、抗鼠标IgG(1:1,000;多克隆;Thermo Fisher,31430)或抗兔IgG(1:1,000;多克隆;Thermo Fisher,A16116)检测初级抗体。使用Zeiss AxioScan幻灯片扫描仪对幻灯片进行成像,并使用QuPath 0.3源软件50进行分析。
对于免疫荧光,石蜡嵌入的载玻片在柠檬酸钠缓冲液(pH 6.0)中煮沸15分钟,然后在阻塞缓冲液中孵化30分钟(PBS中1%的BSA和5%的FCS),并在4°C下用初级抗体孵化过夜。冷冻载玻片在室温下风干,在4%的对甲醛(PFA)中固定10分钟,在SuperBlock溶液(Thermo Fisher)中孵化30分钟,然后用初级抗体孵化1小时。使用的主要抗体是CD3(1:100;Abcam,ab5690)和B220(1:100;BioLegend,RA3-6B2)。幻灯片在PBS中清洗了三次,在室温下用山羊抗兔546(1:200;Thermo Fisher,A-11035)和山羊抗鼠488(1:200;Thermo Fisher,A-11006)在黑暗中孵化1小时,并安装DAPI。幻灯片在蔡司直立710或蔡司AxioScan显微镜上通过共聚焦显微镜成像。
组织清理和三维免疫荧光
组织清理是按照之前描述的51进行的。简而言之,在携带肿瘤的肺部灌注20毫升冷PBS,在10%的中性缓冲福尔马林(Sigma-Aldrich)中固定24小时,并在一夜之间用1:1:4 H2O2:DMSO:PBS脱色。在44°C下,在40毫克毫升-1 SDS和12.36毫克毫升-1硼酸盐中取回抗原后,样品在PBS中用0.2%的Triton X-100洗涤三次,在室温下阻断并孵化48小时,抗体有CD3(1:100;Abcam,ab5690)、B220(1:100;BioLegend,RA3-6B2)和TTF1(1:100;Abcam,ab72876)。样品在PBS中洗涤三次,并在黑暗中孵化48小时,使用荧光标记的抗兔Alexa Fluor 546(1:100;Thermo Fisher,A10040),抗兔Alexa Fluor 546 IgG(1:200;Thermo Fisher,A-11035),抗兔Alexa Fluor 594(1:100;Thermo Fisher,R37119),抗大鼠Alexa Fluor 488(1:100;A-21208),抗大鼠Alexa Fluor 488 IgG(1:200;多克隆;Thermo Fisher,A-11006),抗大鼠Alexa Fluor 647(1:100;Thermo Fisher,A48272),抗老鼠Alexa Fluor 488(1:100;Thermo Fisher,A-21202)或抗山羊Alexa Fluor 647(1:100;Thermo Fisher,A-21447)抗体。样品在PBS中洗涤三次,通过增加的甲醇梯度脱水,并通过增加的水杨酸甲酯梯度清除。清除的样品通过LAvision Ultramicroscope II(Miltenyi)上的光片显微镜或Zeiss Invert 780上的共聚焦显微镜成像,并使用Imaris软件9.8(Bitplane)渲染。
TLS检测和量化
成熟的TLS在这里被定义为存在分离的T细胞和B细胞区域的淋巴聚集体,以及正在进行的GC反应的证据。后者基于GCs中暗区和浅区的区别,在TRACERx(扩展数据图6d)中的诊断性苏木精和伊红染色上确定,或由Ki67染色和小鼠样本中PNA结合的阳性揭示。当TRACERx患者可以使用多个诊断幻灯片时,会汇总TLS计数。在低功率放大时可见,但没有任何GC形成迹象的淋巴细胞簇被认为是淋巴聚集体。
抗体结合和亲和力测定
对于抗体结合,KPAR、KPB6、M. dunni、M. dunni.KARV、HEK293T.ERV3-1env、HEK293T.HERV-K(HML-2)env或HEK293T细胞用热灭活血清或等离子体在室温下在PBS中稀释1:5030分钟,用FACS缓冲液洗涤,在室温下用荧光标记的小鼠或人类IgG、IgA和IgM抗体染色30分钟,并在Ze5分析仪上通过流式细胞仪进行分析。抗体滴度表示为每个抗体同种类型的MFI。对于阻断实验,在染色前,用稀释的血清或血浆在室温下培养10 µg ml-1重组ERVK-7包膜蛋白(Cusabio,CSB-CF351062HU)或H1流感H1N1 HA(鼻生物学,11085-V08H)在室温下培养30分钟。为了检测ERV3-1和HERV-K(HML-2)包膜反应抗体,HEK293T、HEK293T.ERV3-1env和HEK293T.HERV-K(HML-2)env细胞以相等的比例混合,并根据GFP表达水平进行区分(扩展数据图12b)。与亲本HEK293T细胞相比,使用以下公式计算了特定的MFI增加:(GFP+细胞的MFI-GFP-细胞的MFI)/GFP-细胞的MFI,如前所述29。热图是使用Microsoft Excel 2016制作的。对于A549结合,使用以下公式计算了特定的MFI增加:(染色细胞的MFI-无血清控制细胞的MFI)/无血清控制细胞的MFI。
在血清亲和力实验中,用血清在冰上稀释1:501小时,将固定的KPAR细胞孵化,并用FACS缓冲液清洗三次。复制井在37°C下孵化1、2、3、5或10分钟,并在冰上用抗IgG染色30分钟。带有孵化的IgG染色以最大MFI的百分比表示,并被认为与抗体off-rate成正比。
对于补体杀伤试验,KPAR细胞在56°C下用1:10稀释血清,无论是否热失活10分钟或抗KARV包膜(J1KK;内部)。细胞在37°C下孵化3小时,并根据制造商的说明通过乳酸脱氢酶(LDH)释放(Abcam)测量细胞毒性。在微板读卡器(Tecan)上测量了450纳米的光学密度,并归一化为无血清阴性对照和裂解缓冲阳性对照。
对于ADCC检测,A549和NK92细胞以1:1的比例培养,血浆在37°C下稀释1:504小时,并根据制造商的说明通过LDH释放(Abcam)测量细胞毒性。值被归一化为单独对A549细胞的负控制,以及对用裂解缓冲液处理的A549细胞的正控制。
RT-qPCR
使用QIAshredder柱(Qiagen)和RNeasy试剂盒(Qiagen)在同质化后从肺部中提取RNA。使用Maxima First-Strand cDNA合成试剂盒(Thermo Fisher)合成,使用Applied Biosystems Fast SYBR Green(Thermo Fisher)使用以下引物进行qPCR:
Cxcl13:F,5′-CATAGATCGGATTCAAGT;R,TCTTGGTCCAGATCACAA-3′
Hprt, F, 5′-TGACACTGGCAAAACAATGCA; R, GGTCCTTTTCACCAGCAAGCT-3′
使用ΔCT方法将值归一化为Hprt表达式。
ELISA
MaxiSorp板(Thermo Fisher)在4°C下用重组可溶性PD-L1外域(内部)在硼酸盐缓冲盐水中涂覆一夜,并在阻断缓冲液中堵塞1小时(PBS中的5%BSA)。Sera在阻断缓冲液中稀释1:50,在室温下用板孵化1小时,然后用PBS-T进行四次洗涤,用HRP结合的抗鼠IgG(1:1,000;Abcam,ab6728)孵化1小时。板是通过在室温下摇晃5分钟后添加50微升TMB衬底(Thermo Fisher)来开发,然后添加50微升的TMB停止溶液(Thermo Fisher)。在微板读取器(Tecan)上测量了450纳米的光学密度。
单细胞BCR测序和抗体生产
从三只小鼠中汇集的排序活CD45+B220+细胞种群被加载到10倍基因组铬控制器上,并根据制造商的指南准备了VDJ库。使用Illumina HiSeq 2500高输出平台对样品进行了测序。使用10X Genomics CellRanger工作流程执行了脚本对齐和特征-条形码矩阵的生成。
J1KK单克隆抗体作为小鼠IgA或IgG1从显性BCR序列克隆到pRV-IgK-T2A-IgH-IRES-GFP质粒(Genscript)并转化为HEK293T细胞。根据制造商的说明,分别使用蛋白质L自旋柱(Thermo Fisher)和蛋白质A Plus自旋柱(Thermo Fisher)从无血清上清液中纯化IgA和IgG1抗体。
免疫沉淀和质谱法
对于免疫沉淀,根据制造商的说明,J1KK抗体或小鼠IgA同种型对照(Abcam)与Dynabeads(Thermo Fisher)耦合。抗体共轭的Dynabeads随后用从KPAR细胞中收集的4毫克蛋白质化物进行孵化,并在4°C下循环浸泡一夜。使用RIPA缓冲液与蛋白酶和磷酸酶抑制剂鸡尾酒(Roche)一起清洗珠子三次。样品通过在NuPAGE LDS样品缓冲液(Thermo Fisher)中重新悬浮并在95°C下孵化5分钟来洗脱。洗发蛋白在NuPAGE 4-12%的Bis-Tris凝胶(Thermo Fisher)上运行,并使用InstantBlue Coomassie Protein Stain(Abcam)可视化。从每条车道中切除70 kDa的凝胶带,并通过质谱法进行分析。
对于质谱法,切除的蛋白质凝胶片被放置在1.5毫升的Eppendorf管中,用50%(v/v)乙腈和50 mM碳酸氢铵染色,用10 mM二硫代糖醇(DTT)还原,用55 mM碘乙酰胺烷基化。烷基化后,蛋白质在37°C下用6.5纳克μl-1胰蛋白酶(Promega)消化一夜。由此产生的肽在2%(v/v)甲酸、2%(v/v)乙腈中提取,并使用Ultimate U3000 HPLC(Thermo Scientific Dionex)用纳米级毛细管LC-MS/MS进行分析,以提供约250 nl min-1的流速。C18 Acclaim PepMap100 5 μm,100 μm × 20 mm nanoViper 柱(Thermo Scientific Dionex)在 EASY-Spray PepMap RSLC 2 μm、100 Å、75 μm × 500 mm nanoViper 柱(ThermoScientific Dionex)上分离前将肽困住了肽。肽以120分钟的乙腈梯度(2%至80%)被洗脱。分析柱出口通过纳米流电喷雾电离源直接与混合四极Orbitrap质谱仪(Eclipse Orbitrap,ThermoScientific)接口。数据收集在数据依赖采集(DDA)模式下进行,r = 120,000(在m/z 200)从m/z 400-2,000进行全MS扫描,目标AGC值为4×105离子,然后在r = 17,500(m/z 200)时进行20次MS/MS扫描,目标AGC值为1×104离子。使用30的阈值能量收集MS/MS扫描,用于高能碰撞解离(HCD),并使用30秒的动态排除来增加覆盖深度。MS/MS数据使用脚手架软件82(蛋白质组软件)进行验证,并使用1%的虚假发现率(FDR)阈值进行人工审问,以进行蛋白质识别。
TRACERx队列
这项研究的数据是TRACERx队列前瞻性分析的前421名患者的一部分(NCT01888601由伦敦国家研究伦理服务委员会批准,赞助商批准该研究由伦敦大学学院批准,详情如下:REC参考13/LO/1546,协议编号UCL/12/0279,IRAS项目ID 138871)。数据收集遵循了与前100名患者研究中描述的步骤类似的步骤52,53,并在随附的研究54,55,56中进行了全面描述。进入TRACERx研究的知情同意是强制性的,并且是从每个患者那里获得的。
TRACERx RNA-seq队列
本研究中分析的转录组学数据(每个样本5000万配对读数,长度为75 bp或每次读数100 bp)来自TRACERx队列,该队列在随附研究中进行了全面描述54,55,56。数据收集遵循了与之前描述的类似的步骤57。根据病理学和测序分析确定的多个原发性肿瘤患者被排除在外,以避免与多种组织学和/或独立肿瘤谱系相关的潜在混杂变量。仅包括从初步手术切除中提取的原发性和邻近正常肺组织样本的数据,以及图5b中描述的一个淋巴结转移。补充表2总结了研究中分析的TRACERx RNA-seq队列。
HERV转录物识别、读取映射和RNA-seq数据的量化
HERV病毒和其他重复区域如前所述进行了注释58。简而言之,使用使用RepeatMasker(配置nhmmer来注释GRCh38)来注释代表已知人类重复家族的隐藏马尔可夫模型(HMMs)(Dfam 2.0 library v.150923)。RepeatMasker分别注释长终端重复(LTR)和内部区域;因此,对表格输出进行解析,以合并同一元素的相邻注释。汇编了具有功能性env ORF的HERV病毒列表(补充表1),并使用在TCGA的RNA-seq数据子集上组装的自定义转录组对TCGA、GTEx和TRACERx的RNA-seq读数进行了映射和计数,如前所述58。简而言之,使用使用GNU并行和Salmon(v.0.12.0)59的自定义Bash管道计算了成绩单汇编中的所有成绩单的TPM值。然后将TPM值导入Qlucore Omics Explorer v.3.3(Qlucore),用于下游微分表达式分析和可视化。在从给定的HERV原病毒转录的多个转录物的情况下,通过将任何与该原病毒的env ORF重叠的多个转录物的表达相和来折叠数据。患者级平均值在多个原发性肿瘤区域计算,视情况而定。
来自RNA-seq数据的免疫细胞和TLS估计
Danaher等人60的方法用于从肺癌患者的RNA-seq数据中估计免疫细胞种群。患者级平均值在多个原发性肿瘤区域计算,视情况而定。对于小鼠LUAD模型,使用MCPCounter方法61从RNA-seq数据中量化免疫和基质细胞种群丰度。TLS基因集评分是按照之前描述的62计算的。简而言之,TPM值被分位数归一化,并对数转换为log2(值+1)。该评分是根据九个TLS签名基因(CD79B、EIF1AY、PTGDS、RBP5、CCR6、SKAP1、LAT、CETP和CD1D)的平均表达计算的。
来自RNA-seq数据的BCR重建
BCR CDR3序列和类开关使用TRUST4 v.1.0.8开源算法63(https://github.com/liulab-dfci/TRUST4)从RNA-seq BAM文件中组装,并带有默认参数。汇总了编码相同氨基酸(CDR3aa)序列的多个BCR CDR3序列。框架外和部分CDR3序列被排除在外,以仅保留生产序列。多样性被定义为每个样本独特的生产性CDR3aa序列的总数。患者级多样性代表了所有原发性肿瘤区域的独特生产性CDR3aa序列的总数。每个样本的类开关频率计算为分类为IGHM、IGGG、IGHA、IGHE或其他的独特生产CDR3aa序列的比例。患者级平均值在多个原发性肿瘤区域计算,视情况而定。
TRACERx全外显子组测序队列
本研究中分析的全外显子组测序数据(中位深度为413×)来自TRACERx队列,该队列在伴随的研究中得到了充分描述54,55,56。仅包括TP53、EGFR和KRAS中的驱动单核苷酸变体(SNVs)和indels进行分析。对于拷贝数分析,提取了长度>5 bp的段,与ERVK-7位点坐标(GRCh37 chr1:155596185–155606777)有任何重叠,以供分析。为每个样本计算了位点的倍体调整副本数,并使用患者级最大值与转录组学数据相关。TMB是在区域一级通过计算RefSeq定义的非同义编码突变(2014年下载)来计算的,除以所有编码序列的总长度,然后乘以106。
TRACERx血浆队列
根据研究协议,纵向收集患者血浆。在K2 EDTA管中收集了新鲜血液样本。在血液采集后2小时内,使用冷藏离心机在1000克下进行双离心10分钟,然后用2000克10分钟来制备血浆,以去除细胞和血小板。血浆在-80°C下储存在1毫升等份量中。手术前,血浆是在初始手术的前一天或当天收集的(n = 58 LUAD,n = 24 LUSC)。48名LUAD患者和20名LUSC患者有相应的RNA-seq数据;53名LUAD患者有相应的体细胞拷贝数改变数据,而LUSC患者没有对此进行评估。七名患者接受了ICB(nivolumab或atezolizumab),并有治疗血浆可用。CRUK0284患者在组织学上都有LUAD和类癌素生长的明显病变。
TCGA样本的额外生物信息学分析
对于TCGA LUAD样本,之前计算了全球甲基化值的指数64。SOX2表达式,每千基转录物每百万个映射读取上四分位数(FPKM-UQ)的片段,以及ERVK-7基因组位置的平均拷贝数(hg38 chr1:155629344–155634870)从UCSC Xena浏览器65(https://xena.ucsc.edu)下载。
TRACERx、TCGA和SMC队列结果分析
对于TRACERx患者,独立对LUAD和LUSC患者进行了无病生存分析。无病生存期(DFS)被定义为从注册之日起到通过放射学确认注册为TRACERx的原发性肿瘤复发的时间或因任何原因死亡的时间。在随访期间,三名患者(CRUK0512、CRUK0373和CRUK0511)患上了新的原发性癌症,随后从第一原发性肺癌或随访期间诊断出的新原发性癌症复发。由于第三种肿瘤的起源不确定,在诊断新的原发性癌症进行DFS分析时,对这些病例进行了审查。根据组织学特异性队列中位数,将患者级数据分为高组和低组,并使用生存R包(v.3.2.13)的Kaplan-Meier估计值比较了DFS的概率。对于TCGA患者,样本按CXCL13、CD79A、CD19或MS4A1表达式进行排名,并通过对数排名分析比较顶部和底部表达四分位数的生存曲线。对于SMC LUAD队列34的结果分析,样本根据ERVK-7表达式(与该病毒的env ORF重叠的任何多个转录物的总和TPM)进行分层,使用20 TPM的截止值来定义高和低ERVK-7表达。
统计和可重复性
使用GraphPad Prism 7(GraphPad软件)、SigmaPlot 14.0或R(版本3.6.1-4.0.0)进行了统计比较。dplyr(v.1.0.7)、data.table(v.1.14.2)、tidyverse(v.1.3.1)和rjson(v.0.2.20)包用于R中的数据处理。软件包Hmisc(v.4.6.0)用于斯皮尔曼的相关分析。lme4包(v.1.1.27.1)用于线性混合效应模型。套餐生存率(v.3.2.13)用于与患者结果指标的统计关联。通过未配对或配对的学生t测试或单向方差分析与Bonferroni校正进行多项比较,对满足方差标准的正态分布值进行了参数比较。未通过方差测试的数据与非参数双尾曼-惠特尼秩和测试(用于未配对比较)、Wilcoxon签名排名测试(用于配对比较)或带有Tukey或邓恩校正的排名测试的方差分析进行比较,以进行多次比较。使用线性混合效应模型将多区域数据作为随机效应与每个患者进行比较。
报告摘要
有关研究设计的更多信息,请参阅本文链接的自然投资组合报告摘要。
数据可用性
本研究期间使用的RNA-seq和全外显子组测序数据(分别来自TRACERx研究)已存放在欧洲基因组-现象基因组档案馆(EGA)中,该档案馆由欧洲生物信息学研究所(EBI)和基因组控制中心(CRG)托管,加入代码EGAS00001006517(RNA-seq)和EGAS00001006494(全外显子组测序);访问由TRACERx数据访问委员会控制。支持本研究结果的其他数据可在论文及其补充信息中找到。用于本手稿中描述的分析的TCGA和GTEx数据是在2017年从dbGaP(https://dbgap.ncbi.nlm.nih.gov)加入编号phs000178.v10.p8和phs000424.v7.p2中获得的。从UCSC Xena浏览器(https://xena.ucsc.edu)下载了ERVK-7基因组位点的其他TCGA LUAD表达数据和平均拷贝数。核苷酸序列是从NCBI核苷酸资源(https://www.ncbi.nlm.nih.gov/nuccore)下载的。源数据与本文一起提供。
参考文献
- Schumacher, T. N. & Thommen, D. S. Tertiary lymphoid structures in cancer. Science 375, eabf9419 (2022).
- Laumont, C. M., Banville, A. C., Gilardi, M., Hollern, D. P. & Nelson, B. H. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat. Rev. Cancer 22, 414–430 (2022).
- Boumelha, J. et al. An immunogenic model of KRAS-mutant lung cancer enables evaluation of targeted therapy and immunotherapy combinations. Cancer Res. 82, 3435–3448 (2022).
- Hellmann, M. D. et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N. Engl. J. Med. 381, 2020–2031 (2019).
- Patil, N. S. et al. Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell lung cancer. Cancer Cell 40, 289–300 (2022).
- Hao, D. et al. The single-cell immunogenomic landscape of B and plasma cells in early-stage lung adenocarcinoma. Cancer Discov. 12, 2626–2645 (2022).
- Mesin, L., Ersching, J. & Victora, G. D. Germinal center B cell dynamics. Immunity 45, 471–482 (2016).
- Litchfield, K. et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184, 596–614 (2021).
- Leader, A. M. et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 39, 1594–1609 (2021).
- Zhang, Y. et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593 (2021).
- Preuss, K. D., Zwick, C., Bormann, C., Neumann, F. & Pfreundschuh, M. Analysis of the B-cell repertoire against antigens expressed by human neoplasms. Immunol. Rev. 188, 43–50 (2002).
- Kassiotis, G. & Stoye, J. P. Immune responses to endogenous retroelements: taking the bad with the good. Nat. Rev. Immunol. 16, 207–219 (2016).
- Li, M., Huang, X., Zhu, Z. & Gorelik, E. Sequence and insertion sites of murine melanoma-associated retrovirus. J. Virol. 73, 9178–9186 (1999).
- Pothlichet, J., Mangeney, M. & Heidmann, T. Mobility and integration sites of a murine C57BL/6 melanoma endogenous retrovirus involved in tumor progression in vivo. Int. J. Cancer 119, 1869–1877 (2006).
- Ottina, E. et al. Restoration of endogenous retrovirus infectivity impacts mouse cancer models. Cancer Immunol. Res. 6, 1292–1300 (2018).
- Dieu-Nosjean, M. C. et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J. Clin. Oncol. 26, 4410–4417 (2008).
- Germain, C. et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am. J. Respir. Crit. Care Med. 189, 832–844 (2014).
- Vanhersecke, L. et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat. Cancer 2, 794–802 (2021).
- Panova, V., Attig, J., Young, G. R., Stoye, J. P. & Kassiotis, G. Antibody-induced internalisation of retroviral envelope glycoproteins is a signal initiation event. PLoS Pathog. 16, e1008605 (2020).
- Young, G. R. et al. Resurrection of endogenous retroviruses in antibody-deficient mice. Nature 491, 774–778 (2012).
- Shi, J. et al. PD-1 controls follicular T helper cell positioning and function. Immunity 49, 264–274 (2018).
- Good-Jacobson, K. L. et al. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 11, 535–542 (2010).
- Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
- Rowland, S. L., DePersis, C. L., Torres, R. M. & Pelanda, R. Ras activation of Erk restores impaired tonic BCR signaling and rescues immature B cell differentiation. J. Exp. Med. 207, 607–621 (2010).
- Denton, A. E. et al. Type I interferon induces CXCL13 to support ectopic germinal center formation. J. Exp. Med. 216, 621–637 (2019).
- Bindea, G. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795 (2013).
- Ukita, M. et al. CXCL13-producing CD4+ T cells accumulate in the early phase of tertiary lymphoid structures in ovarian cancer. JCI Insight 7, e157215 (2022).
- Kasperek, A. et al. Therapeutic potential of the human endogenous retroviral envelope protein HEMO: a pan-cancer analysis. Mol. Oncol. 16, 1451–1473 (2021).
- Deakin, C. T. et al. Favorable antibody responses to human coronaviruses in children and adolescents with autoimmune rheumatic diseases. Med 2, 1093–1109 (2021).
- Fendler, A. et al. Functional antibody and T cell immunity following SARS-CoV-2 infection, including by variants of concern, in patients with cancer: the CAPTURE study. Nat. Cancer 2, 1321–1337 (2021).
- Monde, K. et al. Movements of ancient human endogenous retroviruses detected in SOX2-expressing cells. J. Virol. 96, e0035622 (2022).
- Watkins, T. B. K. et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 587, 126–132 (2020).
- Wang-Johanning, F. et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J. Natl Cancer Inst. 104, 189–210 (2012).
- Park, S. et al. Artificial intelligence-powered spatial analysis of tumor-infiltrating lymphocytes as complementary biomarker for immune checkpoint inhibition in non-small-cell lung cancer. J. Clin. Oncol. 40, 1916–1928 (2022).
- Hollern, D. P. et al. B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell 179, 1191–1206 (2019).
- Herati, R. S. et al. PD-1 directed immunotherapy alters Tfh and humoral immune responses to seasonal influenza vaccine. Nat. Immunol. 23, 1183–1192 (2022).
- Song, I. H. et al. Predictive value of tertiary lymphoid structures assessed by high endothelial venule counts in the neoadjuvant setting of triple-negative breast cancer. Cancer Res. Treat. 49, 399–407 (2017).
- Lee, H. J. et al. Prognostic significance of tumor-infiltrating lymphocytes and the tertiary lymphoid Structures in HER2-positive breast cancer treated with adjuvant trastuzumab. Am. J. Clin. Pathol. 144, 278–288 (2015).
- Sahin, U. et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc. Natl Acad. Sci. USA 92, 11810–11813 (1995).
- Leong, S. P. et al. Expression and modulation of a retrovirus-associated antigen by murine melanoma cells. Cancer Res. 48, 4954–4958 (1988).
- Kassiotis, G. & Stoye, J. P. Making a virtue of necessity: the pleiotropic role of human endogenous retroviruses in cancer. Philos. Trans. R. Soc. Lond. B 372, 20160277 (2017).
- Wang-Johanning, F. et al. Expression of human endogenous retrovirus K envelope transcripts in human breast cancer. Clin. Cancer Res. 7, 1553–1560 (2001).
- Parikh, A. R. et al. Radiation therapy enhances immunotherapy response in microsatellite stable colorectal and pancreatic adenocarcinoma in a phase II trial. Nat. Cancer 2, 1124–1135 (2021).
- Tokuyama, M. et al. Antibodies against human endogenous retrovirus K102 envelope activate neutrophils in systemic lupus erythematosus. J. Exp. Med. 218, e20191766 (2021).
- Dogan, I. et al. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 10, 1292–1299 (2009).
- Casola, S. et al. Tracking germinal center B cells expressing germ-line immunoglobulin γ1 transcripts by conditional gene targeting. Proc. Natl Acad. Sci. USA 103, 7396 (2006).
- Srinivas, S. et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 (2001).
- Snippert, H. J. et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144 (2010).
- Hanke, K. et al. Reconstitution of the ancestral glycoprotein of human endogenous retrovirus K and modulation of its functional activity by truncation of the cytoplasmic domain. J. Virol. 83, 12790–12800 (2009).
- Bankhead, P. et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
- Messal, H. A. et al. Antigen retrieval and clearing for whole-organ immunofluorescence by FLASH. Nat. Protoc. 16, 239–262 (2021).
- Jamal-Hanjani, M. et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 (2017).
- Bailey, C. et al. Tracking cancer evolution through the disease course. Cancer Discov. 11, 916–932 (2021).
- Al Bakir, M. et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature https://doi.org/10.1038/s41586-023-05729-x (2023).
- Frankell, A. M. et al. The evolution of lung cancer and impact of subclonal selection in TRACERx. Nature https://doi.org/10.1038/s41586-023-05783-5 (2023).
- Martínez-Ruiz, C. et al. Genomic–transcriptomic evolution in lung cancer and metastasis. Nature https://doi.org/10.1038/s41586-023-05706-4 (2023).
- Rosenthal, R. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019).
- Attig, J. et al. LTR retroelement expansion of the human cancer transcriptome and immunopeptidome revealed by de novo transcript assembly. Genome Res. 29, 1578–1590 (2019).
- Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
- Danaher, P. et al. Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer 5, 18 (2017).
- Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016).
- Cabrita, R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020).
- Song, L. et al. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat. Methods 18, 627–630 (2021).
- Jung, H. et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat. Commun. 10, 4278 (2019).
- Goldman, M. J. et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 38, 675–678 (2020)